Aceruloplasminemia is an inherited genetic disorder which is caused by defects in the ceruloplasmin gene. Aceruloplasminemia is characterised by diabetes, retinal degradation and progressive neurogical symptoms, including extrapyramidal syndrom, ataxia and dementia [1]. Recent papers [1-3] associating ceruloplasmin deficiency with aceruloplasminemia and severe iron deposition in visceral organs and brain tissues have focused attention on the role of ceruloplasmin as ferroxidase in human plasma (Fig. 1).

Ceruloplasmin is initially synthesized as apoceruloplasmin and incorporates copper into the apoprotein in the Golgy body, resulting in the formation of holoceruloplasmin prior to extracellular secretion. In normal subjects, iron is continuously recycled between bone marrow and hepatocytes with serum transferrin acting as shuttle to deliver iron from hepatocytes to the bone marrow. The role of ceruloplasmin, formed as holoceruloplasmin is a ferroxidase mediating ferrous ion oxidation and subsequent transfer to transferrin. In normal subjects copper enters the cell and binds the copper chaperons which deliver copper to ATP7B. The ATP7B pumps the copper into the trans-Golgi network
Mutant ceruloplasmin biosynthesis were investigated using cell culture system without endogenous ceruloplasmin [2]. The mutant protein revealed three distinct pathological mechanisms including mutants associated with mistrafficking, resulting in the retention of the protein in the ER, mutants altering the intrinsic protein structure, resulting in abrogation of copper incorporation into apoceruloplasmin and mutants with impaired ferroxidase activity (Fig. 1,2)

Missense mutations causing aceruloplasminemia were investigated by testing their ability to prevent ferroportin degradation in rat glioma C6 cells silenced for endogenous ceruloplasmin [3]. Most of the mutants did not complement (i.e. did not stabilize ferroportin) because of the irreversible loss of copper binding ability. Mutant R701W,which was found in a heterozygous very young patient with severe neurological problems,was unable to complement per se, but did so in the presence of copper-glutathione or when the yeast copper ATPase Ccc2p was co-expressed,indicating that the protein was structurally able to bind copper, but that metal loading involving the mammalian copper ATPase ATP7B was impaired. Notably,R701W exerted a dominant negative effect on wild type,and it induced the subcellular relocalization of ATP7B. These results constitute the first evidence of “functional silencing”of ATP7B as a novel molecular defect in aceruloplasminemia.
To elucidate the molecular pathogenesis of aceruloplasminemia, the biosynthesis of a missense mutant ceruloplasmin (P177R) occurring in an affected patient was examined [4]. Chinese hamster ovary cells transfected with cDNAs encoding secreted and glycosylphosphatidylinositol (GPI)-linked wild-type or P177R human ceruloplasmin were examined by pulse chase metabolic labeling. These experiments, as well as immunofluorescent analysis and N-linked glycosylation studies, indicate that both the secreted and GPI-linked forms of the P177R mutant are retained in the endoplasmic reticulum (ER). The P177R mutation resides within a motif, which is repeated six times in human ceruloplasmin and is conserved in the homologous proteins hephaestin and factor VIII. Analysis of additional mutations in these motifs suggests a critical role for this region in ceruloplasmin trafficking and indicates that substitution of the arginine residue is critical to the ER retention of the P177R mutant. Metabolic labeling of transfected Chinese hamster ovary cells with 64-Cu indicates that the P177R mutant is retained in the ER as an apoprotein and that copper is incorporated into both secreted and GPI-linked ceruloplasmin as a late event in the secretory pathway.
———————–
In certain cases (Fig. 2), mutations in the ceruloplasmin gene with aceruloplasminemia appear to be caused by result in an incomplete expression of the ceruloplasmin molecule, with the polypeptide chain being prematurely terminated [2,5]. Our earlier work on the X-ray structure of human ceruloplasmin (PDB ID: 1kcw ) provided structural explanations as to why, if the truncated molecules are actually secreted into the bloodstream, they will have their oxidase function severely impaired [5,6]. Thus, Fig. 3 shows the trace of the a-carbon polypeptide backbone for domains 1 and 6 of human ceruloplasmin only; domain 1 is shaded red on the left hand side of the figure. The blue and magenta structures on the right hand side of the figure represent domain 6, and, in particular, the blue trace is the portion of the polypeptide chain, residues 991 to 1046 inclusive, which would be missing if incorrect expression terminated the chain at 991 [5]. The copper-binding ligands that would be concomitantly missing comprise three of the ligands that bind to the domain 6 mononuclear copper (C1021, H1026 and M1031). In the absence of these ligands, it can be concluded that domain 6 is very unlikely to bind copper, and this should have a profound effect on the oxidase efficiency of the enzyme. In addition, two of the histidine residues that bind to the trinuclear cluster (H1020 and H1022) are also absent, which gives rise to the possibilities that this cluster may not bind in the same way as in the intact enzyme or that it may not bind at all; in either case this will affect the oxidase activity of the enzyme. Further, the amino acid residues on the missing peptide contribute over 50% of the hydrogen-bond and ion-pair interactions between domains 1 and 6 in the intact enzyme. Such a configuration will be even more susceptible to proteolytic cleavage than the correctly folded enzyme. The net effect of the missing C-terminal portion of the polypeptide chain is that the enzyme may not fold into the organisational tertiary structure depicted in Fig. 3 and that its ability to act as an oxidase will be severely impaired with respect to the intact enzyme. Incomplete folding and increased susceptibility to proteolytic cleavage would be consistent with the observed aceruloplasminemia such as the failure to find ceruloplasmin in liver extracts by the Western blot and ELISA methods [5]. An impairment of the ferroxidase activity would be consistent with a gradual accumulation of intracellular iron leading to systemic haemosiderosis; the truncated enzyme would not mediate the release of iron from cells nor its oxidation prior to uptake by transferrin. .

Structural studies of ceruloplasmin mutants
I have selected a few mutations in the wild type human ceruloplasmin structure (PDB ID: 2j5w, [7]) from the list above (Fig. 1,2) for my studies: three missense mutations, G631R, G969S and H978Q and two nonsense mutations W1017X and R882X. I have been using the web portal Missense3D [8] for the analysis of structural damaging change in the mutant structure. An in-house pipeline, Missense3D, for structurally assessing missense variants was made available at http://www.sbg.bio.ic.ac.uk/~missense3d/ (Fig. 4)
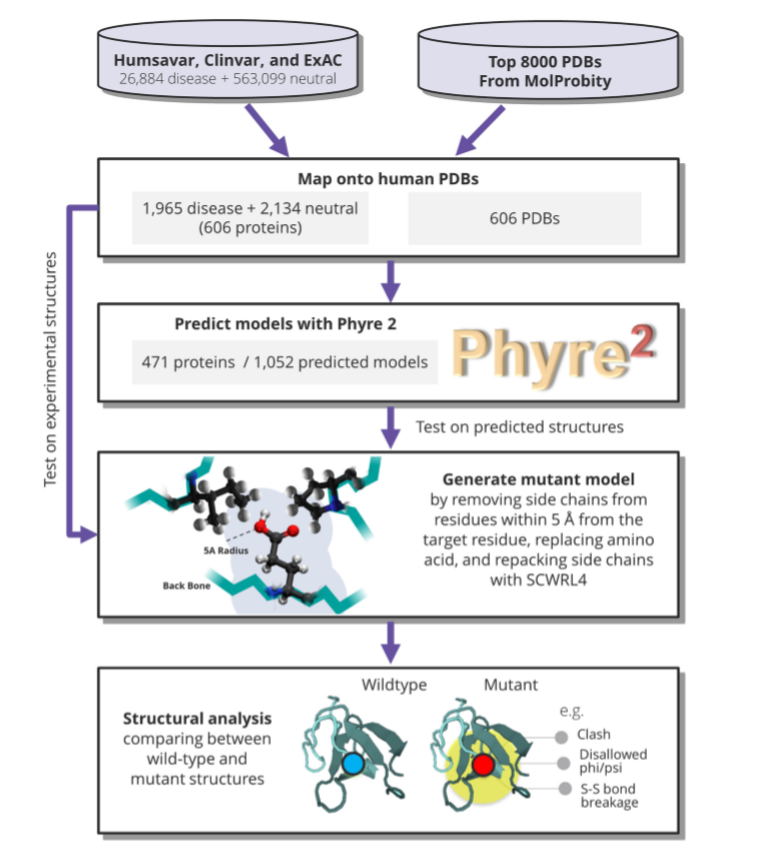
PHYRE2 applies hidden Markov chain algorithm [9] to predict target mutant structure using template provided by the user. Accurate side chain rotamer conformation for a mutated residue is achieved in a program SCWRL4 [10] through: (1) a new backbone-dependent rotamer library based on kernel density estimates; (2) averaging over samples of conformations about the positions in the rotamer library; (3) a fast anisotropic hydrogen bonding function; (4) a short-range, soft van der Waals atom-atom interaction potential; (5) fast collision detection using k-discrete oriented polytopes; (6) a tree decomposition algorithm to solve the combinatorial problem; and (7) optimization of all parameters by determining the interaction graph within the crystal environment using symmetry operators of the crystallographic space group.
Missense mutations, results
1. G631R
This substitution replaces a buried uncharged residue GLY with a charged residue ARG (Fig. 5). It also triggers disallowed phi/psi alert. The phi/psi angles are in favored region for wild-type residue but outlier region for mutant residue.
Arg631 of the mutant forms a new salt bridge with Asp635: NH2Agr631 —OD2Asp635. This aspartate is hydrogen bonded to the ligand of the mononuclear copper of the 4th domain, His637 , in the wild-type structure. In addition, Asp635 is one of the conserved residues of the ET pathway between two mononuclear copper centres – Cu2- Cu4: Cu2-H324 …. E633 A634 D635 V636 …. H637-Cu4 [6, 11].
This mutant, together with four others (Q692K, M966V,G969, R882X), is reported as a mutant with impaired copper incorporation (Fig. 1,2). We can suggest that the mutant salt bridge NH2Agr631 —OD2Asp635 affects (weakens) hydrogen bond between OD2 atom of D635 and NE2 atom of His637, one of the ligands of CuT1, and this may trigger the copper loss and/or ability to bind copper in domain 4.

2. G969S
This substitution replaces a buried GLY residue with a buried SER residue and, as in the previous case, it triggers disallowed phi/psi alert . Also, the substitution leads to the expansion of cavity volume by 4.32 Å^3.
Ser969 of the mutant makes a new hydrogen bond with Asp973: OGSer969 —OD1Asp973. This aspartate, in turn, is hydrogen bonded to the ligand of the mononuclear copper of the 6th domain, His975 , in the wild-type structure. Also, Asp973 is one of the conserved residues of the ET pathway between two mononuclear copper centres – Cu4-Cu6: …. E971 I972 D973 L974 …. H975-Cu6 [6, 11].
This mutant is in the list of mutants with impaired copper incorporation. Again, like in previous case with the G631R mutant, we can suggest that formation of a new H-bond in the mutant structure of G969S would affect the copper binding ability in the mononuclear copper centre of domain 6 via the interaction of Ser969 with Asp973. The latter residue is having a structural role to support side chain conformation of the histidine ligand of copper, H975.
.

3. H978Q
Here, for the mutation of the histidine ligand of T2 copper of the trinuclear cluster we observe a bit more dramatic structural damage than in the two previous cases (Fig. 7a,b,c). This substitution replaces a buried charged residue HIS with an uncharged residue GLN and it disrupts all side-chain / side-chain H-bond(s) and/or side-chain / main-chain bond(s) H-bonds formed by a buried HIS residue. Wild-type salt bridge is detected between ND1 atom of HIS 978 and OD1 atom of ASP 995 (distance: 3.783 Å), and the substitution disrupts this bond. Also, the substitution leads to the expansion of cavity volume by 77.976 Å^3.
I suppose, if we could have managed to crystallize this H978Q mutant, we may have observed the CuT2-depleted form of hCP on our electron density maps! It is clearly seen on Fig.7a that copper T2 is weakly/loosely bound in this mutant: charged histidine ligand is replaced by uncharged glutamine. In addition, a new conformation of the second histidine ligand of CuT2, H101, makes NE2H101 —Cu bond much weaker compared to the wild-type bond.
The H978Q mutant is reported as a mutant with no ferroxidase activity (Fig. 1,2). Although the oxygen molecule may be bound at the antiferromagnetically coupled CuT3a-CuT3b cluster, the absense of CuT2 in the trinuclear centre would disrupt the catalytic cycle – for details see pages ‘Ceruloplasmin’ and ‘Laccase’ of this website. Some new experimental evidences ( preferably by the X-ray diffraction methods) are necessary to support the model shown on Fig. 7a.

Rotation of imidazole ring for H101,H103,H980,H1020 ligands and a significant shift of imidazole of H1022, ligand of T3 copper, were observed in the H978Q mutant structure (Fig. 7b). H1022 is no more a ligand in the mutant and this may affect binding of T3 copper.

Earlier, I have created a model of H978Q mutant using SWISS-MODEL server [12] which is freely available at https://swissmodel.expasy.org . I used 2j5w hCP structure as a template with the replacement His978/Gln. All heteroatoms including coppers of the active sites and oxygen molecule were present in the template. Interestingly, although the histidine ligands were in the same place in the mutant structure (imidazole rings were not shifted or rotated), the resulting refined mutant structure did not contain T2 copper and bound oxygen molecule. This area with missing T2 copper and O2 is shown in orange dots on Fig. 7c. Clever programme, indeed! :-). It seems, the bonds from NH2- and =O groups of the Gln978 side chain did not satisfy the criteria of the programme. This again indicates, although it is not supported experimentally, that T2 copper may have been lost in this mutant.

The X-ray structure of the H99Q mutant of small Ssl1 laccase [13] with depleted type-2 copper ion , PDB entry code 4UAN , supports my suggestion that the T2 copper could be missing in the H978Q mutant of hCP.
Together with ceruloplasmin the Ssl1 laccase belongs to the MCO family with the T1-T2/T3 unit of copper-containing active sites conserved for all members of the family (see page ‘Laccase’ in this website).
H99 is one of the ligands of T2 copper in the trinuclear cluster of the Ssl1 laccase. The second ligand is His231. H99 and H231 are structural equivalents of H101 and H978 ligands of T2 copper in hCP. The replacement His99 /Gln in Ssl1 laccase leads to the removal of T2 copper from the protein molecule. Details of the structure could be found in here https://www.wwpdb.org/pdb?id=pdb_00004uan
Nonsense mutations, results
1. W1017X
Expression would be terminated at Trp1017 for this mutant; therefore the portion of the polypeptide chain, residues 1017 to 1041 inclusive, would be missing in the hCP molecule. This missing C-terminal segment (shown in yellow on Fig.8) contains three ligands of the mononuclear copper centre of the 6th domain, C1021, H1026, M1031, one ligand of the labile copper site, D1025, and two histidine ligands of CuT3 of the trinuclear cluster, H1020 and H1022 (Figs 9,10). All these ligands would be missing. For those, who can read and understand Russian: это просто означает, что “все, что было – позабыла/ все, что будет, – позабудет…” ( из песни “Цыганка”, автор Новелла Матвеева) :-).
This mutant is in the list of mutants retained in the endoplasmic reticulum (Fig. 1,2). We can suggest that the loss of the binding ability of copper due to the absence of three ligands in the 6th domain mononuclear Cu centre and two ligands in the trinuclear cluster might have a dramatic impact on the overal conformation of the polypeptide chain. It is probable that the incomplete enzyme will adopt a different organisation of the tertiary structure and may well exist in an open configuration, which would prevent the protein from secretion into the bloodstream.


2. R882X
Expression would be terminated at Arg882 for this mutant; therefore the whole domain 6 would be truncated in the hCP molecule; this C-terminal missing segment of the polypeptide chain is shown in yellow on Fig. 11. Domain 6 of the wild-type protein contains four ligands of the CuT1 centre – H975, C1021, H1026,M1031, two ligands of the labile copper site, H940, D1025 (Fig 12) and four histidine ligands of the trinuclear cluster – H978,H980, H1020 and H1022 (Fig. 13).
Compared to the W1017X mutant, here we could expect more dramatic, even catastrophic, impact on the hCP structure. This mutant is reported as a mutant with impaired copper incorporation. It is not surprising that the CuT1 is not bound in domain 6 when all the ligands of this mononuclear copper site are missing in the truncated mutant enzyme. There is no reason to bind copper in this case (Fig.12) :-). The absence of four histidine ligands of the thinuclear cluster (Fig. 13) means that T2/T3 copper centre would not be properly formed. Interestingly, this mutant is not retained in the ER, like W1017X mutant. The secretion of this protein into the bloodstream would result in complete loss of ferroxidase activity [6,11] and, since the molecular fold would probably be far more open than as shown in Fig. 3 and Fig. 11, the protein would be even more susceptible to proteolytic degradation.


Concluding remarks
All 5 mutant structures shown above, have their mutated residues in a close proximity to the copper atoms of the active sites. This facilitates the analysis – here we could provide some plausible explanation for a mutant malfunction. This is especially true for the nonsense mutations resulting in a truncated enzyme with the copper ligands partly or completely missing.
I am currently trying to work out the other mutants from the aceruloplasminemia list. Obviously, there are limitations in a direct structural approach for the interpretation of the mutants’ dysfunction. Missense3D is not detecting structural damage for some mutants and in many cases graphics (comparative visual analysis of the wild-type and mutant static structures) does not give any clue for the explanation as to why a mutant is inactive.
For example, the mutants P177R and R701W are still mysterious. P177R is the mutant retained in ER, R701W is the mutant with low ferroxidase activity.
P177R: buried proline 177 residue lies in relative proximity to several neighboring hydrophobic residues. It is not clear how the replacement of the uncharged proline by charged arginine in this mutant may lead to the protein unfolding [4]. The entropy cost of this replacement might be high, but this would certainly need further substantiation.
R701W: arginine 701 residue is located on the surface of the molecule in a large solvent-exposed loop connecting domains 4 and 5 and comprising residues 700–708. No structural damage has been detected by the programme Missense3D for the replacement Arg –>Trp. However, according to [3], this mutant belongs to the category of ‘functionally incompetent’, its ferroxidase activity is seriously reduced. The authors [3] suggest that this mutation affects the interaction of ceruloplasmin with ATPase, ATP7B: “the dominant negative effect of hCp R701W could then be due to abnormal interaction of the transporter with the ferroxidase, which could possibly make ATP7B unavailable even for hCpWT”. And further –> “we believe that failure to incorporate copper in WT Cp together with aberrant localization of ATP7B and significant fragmentation of the Golgi apparatus are critical determinants of the severity of the phenotype observed in the heterozygous patient carrying the Cp R701W mutation” [3].
References
1. M.Kruger. The neuropathology and neurodegeneration with brain iron accumulation (2013). Int. Rev. Neurobiology, Ch. 8 ,110:165-194
2. S.Kono (2013). Aceruloplasminenia: an update. Int. Rev. Neurobiology, Ch.6 110: 125-151
3. M.diPatti, N.Maio, G.Rizzo, G.De Francesco, T.Persichini, M.Colasanti, F.Polticelli,and G.Musci (2009). Dominant mutants of ceruloplasmin impair the copper loading machinery in aceruloplasminemia. J.Biol.Chem., 284 (7): 4545-4554
4. N. Hellman, S.Kono, H. Miyajima, and J.D. Gitlin (2002). Biochemical analysis of a missense mutation in aceruloplasminemia. J.Biol.Chem. 277 (2): 1375-1380
5. K.Yoshida, K.Furihata, S.Takeda, A.Nakamura, K.Yamamoto, H.Morita, S.Hiyamuta, S.Ikeda, N.Shimizu, N.Yanagisawa. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans (1995). Nature Genetics. 9 (3): 267–72.
6. P.Lindley, G.Card, I.Zaitseva,V.Zaitsev, B.Reinhammer, E.Selin-Lindgren, K.Yoshida. An X-ray structural study of human ceruloplasmin in relation to ferroxidase activity (1997), J.Biol.Inorg.Chem. 2: 454-463
7. I.Bento,C.Peixoto,V.Zaitsev and P.Lindley. Ceruloplasmin revisited: structural and functional roles of various metal cation-binding sites (2007). Acta Cryst. D63(2): 24O-248
8. S.Ittisoponpisan, S.A.Islam, T.Khanna, E.Alhuzimi, A.David, and M.Sternberg (2019)
Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol., 431: 2197-2212
9. L.Kelley, S. Mezulis ,C.Yates, M.Wass,and M.Sternberg (2015) The Phyre2 web portal for protein modelling, prediction and analysis. Nature Protocols, 10: 845-858
10. G. Krivov, M. Shapovalov, and R. Dunbrack Jr (2009). Improved prediction of protein side-chain conformations with SCWRL4. Proteins, 77: 778–795.
11. P. Lindley, G.Card, I.Zaitseva, V.Zaitsev (1999). Ceruloplasmin: the beginning of the end of an enigma. In: Perspectives of Bioinorganic Chemistry, 4 : 51-82
12. A.Waterhouse, M.Bertoni, S.Bienert, G.Studer, G.Tauriello, R.Gumienny, F.Heer, T.deBeer, C.Rempfer, L.Bordoli, R.Lepore, T.Swede (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46:W1, W296–W303
13. M. Gunne, A.Hoppner, P-L. Hagedoorn and V. Urlacher (2014). Structural and redox properties of the small laccase Ssl1 from Streptomyces sviceus. FEBS Journal, 281:4307–4318
